
Dastidar Lab
Neurodegeneration and Neurotherapeutics Co-laboratory

Research
Neurodegeneration and Neurotherapeutics Co-laboratory
Our long term research interests involve understanding key regulatory pathways and how alterations in gene expression contribute to human aging, age related disorders and infectious diseases. To achieve this, we identify novel genetic targets that might play a role in diseases and disorders. Thereafter, we study the physiological role of the target genes with the ultimate goal of designing synthetic or natural drugs to modulate these targets/pathways. The lab utilizes novel molecular and cell biology tools and techniques, biochemistry, in vitro model systems, MEFs, mice genetics, microbiology and CRISPR/Cas9 based functional genomics screening approaches.
We are currently focusing on the following projects:
Role of Reduced protein translation in neurodegenerative disorder
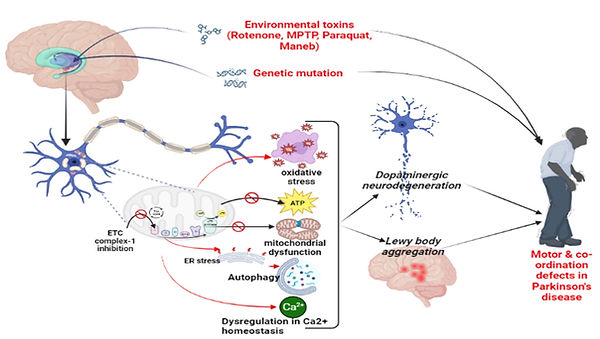
Proteostasis is essential for optimal cell growth and survival, dysregulation of which causes protein aggregation and is associated with age-related neurodegenerative diseases such as Parkinson’s disease (PD). mTOR, a master regulator of proteostasis, is dysregulated in many diseases, such as PD. A key downstream substrate of the mTORC1 complex, eukaryotic translation initiation factor 4E binding protein (4EBP1), has been linked to enhanced longevity in invertebrates. Active 4EBP1, by sequestering eIF4E from eIF4G, blocks 5’ Cap dependent mRNA translation. As reduced protein synthesis may decrease stress on cellular protein quality control systems in the cell, the overall goal of this project is to examine the role of reduced protein translation in neuroprotection, by testing if increased expression of 4EBP1 can ameliorate toxicity in a series of in vitro and in vivo models of PD.
The outcome from this project will not only confirm the importance of reduced protein translation in neuroprotection, but, if successful, will also seek to establish the mechanistic pathways underlying neuroprotection promoted by reduced protein translation. This work will thus have therapeutic potential for debilitating neurodegenerative disorders and possibly other mitochondrial diseases.
To identify activators and repressors of eukaryotic Initiation Factor 4E Binding Protein as potential therapeutic targets
Aberrant mTOR phosphorylation and its downstream effectors has been implicated in many malignancies such as neuroblastoma, glioblastoma multiforme, astrocytoma among many other forms of cancer; age related neurodegenerative disorders such as Parkinson’s Disease; and neurodevelopment disorders such as autism spectrum disorders. So a clear understanding of the downstream effectors of the mTOR pathway is quintessential to explore avenues to improve health as we age and combat age related disorders. We are focusing on 4E-BPs which are downstream effectors of mTOR pathway. The overarching goal of this project is the biological characterization of the 4E-BPs with a focus on identifying their upstream activators or repressors which may represent drugable targets for treatment in cancer of the CNS and neuroinflammatory disorders. This work will thus have therapeutic implications for developing treatments for PD and possibly other pathologically similar neuroinflammatory disorders.

Evaluating the potential of Antisense Oligonucleotides against FUS pathology as a treatment option for Amyotrophic Lateral Sclerosis
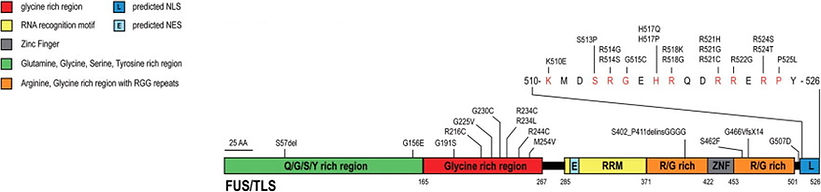
Frontotemporal dementia (FTD) is the second most common dementia after Alzheimer’s disease. Approximately 30% of patients with Amyotrophic Lateral Sclerosis (ALS) develop FTD and show a poor prognosis. Due to overlapping clinical/pathological/biochemical/genetic determinants, it is recognized as the FTD-ALS disease spectrum. Mutations in C9orf72/TARDBP/FUS/TBK1/VCP/SQSTM1 are associated with FTD-ALS. As part of an ongoing ICMR project, we are working on four novel mutations in the FUS gene in ALS patients through collaborations with the Taneja Lab at Sri Gangaram Hospital, New Delhi. In this project, we are targeting FUS with antisense oligonucleotides (ASOs) as a therapeutic strategy. The project aims to work with patient derived iPSC, animal models and in vitro model systems.
Characterising a novel potential antidepressant drug that activates the RXFP3 receptor.

RXFP3 (Relaxin Family Peptide 3 Receptor) is a neurotransmitter receptor expressed within the brain, and is an exciting target for the development of novel antidepressant drugs. For example, rodent studies have shown that drugs which active RXFP3 (agonists) reduce anxiety and display other antidepressant-like actions. However, these early versions of RXFP3 agonists have required direct brain injection, as they do not cross the blood-brain barrier (BBB). This approach is associated with a range of problems, such as the need for surgical implantation of a guide cannula and uneven drug distribution within the brain. Therefore, the crucial next step is to develop and characterize an RXFP3 agonist that crosses the BBB and demonstrates its antidepressant actions following peripheral injection. To achieve this goal, this project will collaborate with world leaders in the RXFP3 field, allowing access to recently developed BBB-penetrating RXFP3 agonists that display exciting in vitro pharmacological properties. This project will screen for RXFP3 agonists and determine which elicits the most promising acute changes in depression-relevant behaviors in mice and rats. The pharmacological specificity of this best-performing RXFP3 agonist will be confirmed using RXFP3 knock-out mice, as on-target RXFP3 drug effects will not be present within this strain of mouse. Importantly, we will also determine the therapeutic potential of chronic RXFP3 agonist treatment in a mouse model of depression, and the acute and chronic mechanisms of action will be characterized via whole transcriptome sequencing of key brain regions post-treatment. Taken together, this project will help validate the therapeutic potential of RXFP3 and will help fast-track the development of an RXFP3 agonist for the treatment of depression by laying the foundation for future clinical trials.
Role of mini-chaperones in alleviating the molecular pathophysiology of Amyotrophic Lateral Sclerosis

Accumulation of misfolded protein aggregates in the CNS is a common feature of many neurodegenerative diseases such as Amyotrophic Lateral Sclerosis (ALS). A unifying mechanism of neurodegeneration in these diseases is unregulated protein aggregation & impaired protein clearance via the autophagy-lysosomal pathway (ALP), which results in disruption of protein homeostasis. This unwanted toxic protein aggregation leads to disruption of normal cellular functions, oxidative stress, nucleo-cytoplasmic transport defect, disruption in RNA processing, protein translation, mitochondrial dysfunction, nucleolar stress and ultimately neuronal death. ALS is a neurodegenerative disease characterized by progressive loss of motor neurons in the brain and spinal cord. Small molecules, peptides, molecular chaperones and antisense oligos are currently being pursued worldwide to address the issues that arise due to protein misfolding and aggregation. We have obtained a patentable, cell-penetrable caspase-3 activable, protease-resistant mini-chaperones which have anti apoptotic effects in in vitro models of cytotoxicity and in AD. We propose to use established in vitro C9ORF72 hexanucleotide repeat-associated dipeptide neuronal toxicity models of ALS to check the efficiency of the mini chaperones in reducing neuronal toxicity as an effective therapeutic molecule for ALS.
Understanding the molecular mechanisms behind the pathophysiology associated with Neurological Melioidosis using in vitro and in vivo model systems.
Burkholderia pseudomallei, a Gram-negative soil saprophyte, causes Melioidosis, an emerging tropical disease. Its genome reveals diverse genes encoding virulence factors that manipulate host processes. This study aims to understand the molecular mechanisms of cellular stress response due to B. pseudomallei infection, focusing on physiological and genetic diversity, virulence, and host-pathogen interaction. Our work mainly aims to characterize Indian clinical isolates of B. pseudomallei, both at the phenotype level and geneotype level and to study genetic diversity among Indian clinical isolates in comparison to global reference strains through bioinformatic analysis. Our work is currently focused on predicting and characterizing Genome Islands (GIs) in several Indian clinical isolates of B. pseudomallei, thereby identifying and characterize potential outer membrane proteins that could be a potential vaccine candidate. We are currently developing in-vito and in-vivo model systems to study the molecular underpinnings employed by the bacterium within the host physiological niche.
